
The software life cycle is long, often slow and tangled across all its phases. The life cycle of software as a medical device (SaMD) and software within medical devices (SiMD) is even more difficult to handle because of the strict regulations that make development workflows necessitate deep investigation.
Specifically, the IEC 62304 is a pivotal standard because it defines all the requirements that characterize the SaMD life cycle, marking the first key step to address compliance.
Luckily, this standard is harmonized internationally, so developers can take it as a unique reference to meet diverse requirements of different global markets.
Here follows a thorough guide with the most asked and discussed questions on the topic: Let’s delve into details of the IEC 62304.
General FAQs About IEC 62304
1. What is IEC 62304?
IEC 62304 is the international standard from the International Electrotechnical Commission (IEC), “Medical device software – Software life cycle processes”. It defines the life cycle requirements (processes, activities, and tasks) necessary for the safe design and maintenance of medical device software. It applies when software is itself a medical device (SaMD) or an embedded/integral part of one (SiMD).
2. How does IEC 62304 compare to other medical device standards (such as ISO 13485, IEC 82304-1, IEC 81001-5-1, ISO 14971, IEC 62366-1)?
IEC 62304 works hand in hand with other standards that cover complementary functions. ISO 13485 sets the Quality Management System for Medical Device manufacturers. IEC 62304 extends ISO 14971 to integrate Risk Management into the SDLC. ISO 14971 addresses risk for the entire Medical Device. IEC 82304-1 applies only to healthcare software products and refers to the processes defined in IEC 62304, which applies to both SaMD and SiMD. IEC 81001-5-1 and IEC 62366-1 handle, respectively, cybersecurity and usability.
3. Is there an IEC 62304 certification?
The IEC itself does not provide certification or any attestation of conformity. Compliance is determined by inspecting the required documentation during ISO 13485 audits, and assessing the processes, activities, and tasks performed based on the software safety class.
Risk Management [Clauses 4.2 and 7]
4. How to implement the risk management process required by the IEC 62304?
The manufacturer must apply a risk management process compliant with ISO 14971. The software risk management process (Clause 7) must be embedded in the device risk management process of ISO 14971. Clause 7 provides additional requirements for risk control specific to software, such as identifying potential software causes contributing to hazardous situations.
5. What is the “Probability of Failure” defined in IEC 62304?
Establishing the Software Safety Classification requires thorough consideration of risks. Such risks come from merging the probability of occurrence of harm and the severity of that harm according to ISO 14971.
6. What is the impact of the “100% Probability of Failure” rule?
The rule applies to the probability of software failure leading from a hazard to an hazardous situation: this probability P1 must be considered 100% (or 1), since no agreement exists on quantitatively measuring software failure odds. This worst-case estimate fits when software contributes to a sequence of events creating a hazardous situation. Conversely, the chance of harm P2 occurring from that hazardous situation can be less than 100%. Please note that the overall probability of harm from software failure is not set to 100%, so risk assessment and subsequent software safety classification emphasize the resulting harm severity instead.
Software Safety Classification [Clause 4.3]
7. What is the IEC 62304 software safety classification?
It classifies software into three types (A, B, or C) based on the worst-case risk of harm resulting from a hazardous situation the software could contribute to. Class A implies no injury or damage to health is possible; Class B, non-serious injury possible; and Class C, death or serious injury possible.
8. What is the correlation between Software Safety Classification and Medical Device Class for EU MDR or Level of Concern for FDA classification?
The two classifications serve distinct purposes and do not have a direct one-to-one correlation. The Medical Device Class (MDR for Europe and FDA for US) sets the bar for regulatory submission, based on the device’s overall risk to the patient and the level of regulatory control needed. On the other hand, the Software Safety Classification decides the level of development rigor required during the SDLC to ensure software safety. In essence, Software Safety Classification focuses on the SDLC especially with regard to software, which is one part of the Medical Device.
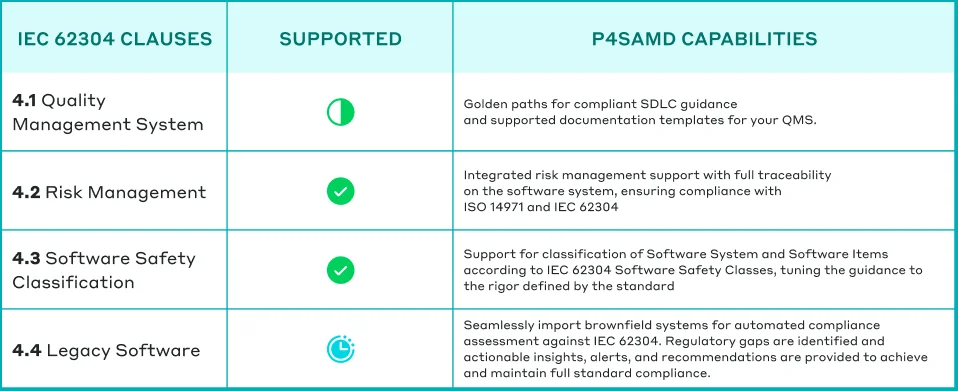
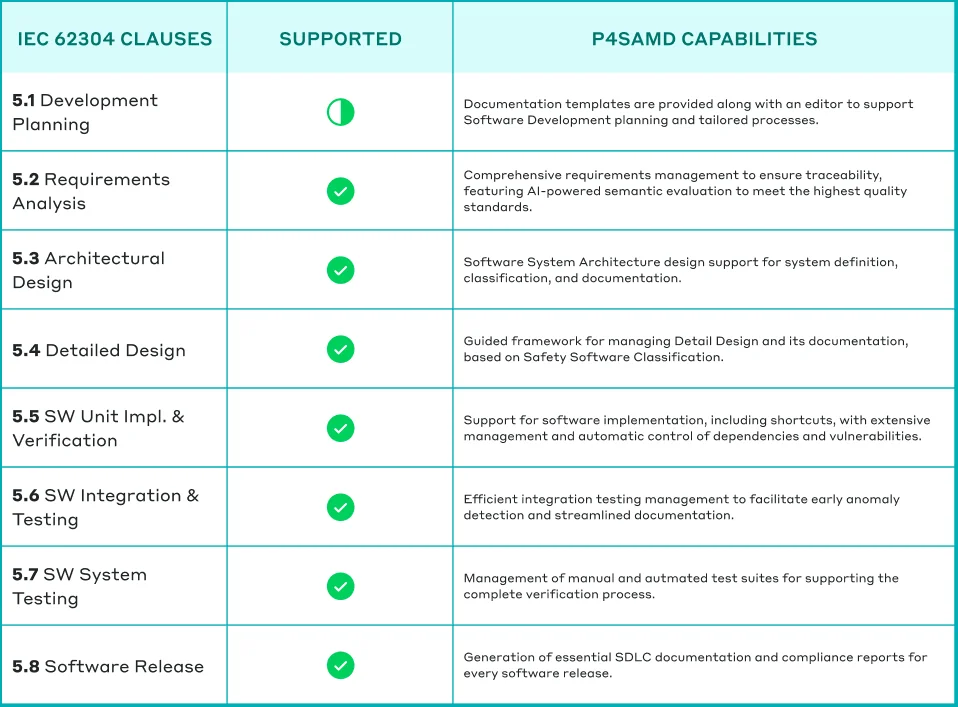
Software Development Process and Planning [Clause 5.1]
9. Does IEC 62304 mandate a specific software development methodology (e.g., Waterfall or Agile)?
No, the standard does not prescribe a specific life cycle model; the manufacturer is responsible for selecting one, such as Waterfall, Incremental, or Evolutionary. The manufacturer maps the required processes, activities, and tasks defined by the standard onto the chosen model.
Requirements Analysis [Clause 5.2]
10. What is the role of the Software Traceability Matrix in demonstrating compliance with IEC 62304?
The traceability records establish the necessary traceability throughout the life cycle. This demonstrates the relationships between system requirements, software requirements, software system tests, and risk control measures implemented in software. This link ensures verification covers all safety and functional requirements.
11. Which information is mandatory for the requirement analysis to be tracked?
Requirements analysis verification must confirm that software requirements implement system/risk control requirements, are non-contradictory, and are unambiguously expressed. Crucially, requirements must be stated such that test criteria can be established, be uniquely identified, and be traceable to their source.
12. What’s the key difference between a functional requirement and a safety requirement?
Functional requirements define the expected capabilities and performance of the software (e.g., timing, characteristics, inputs/outputs). Safety requirements are specifically the risk control measures defined during the risk management process that must be included in the software requirements to control identified risks.
Software Design [Clause 5.3, 5.4]
13. What’s the scope of Architectural Design?
The scope is defining the major structural components of the software and transforming requirements into a documented architecture that identifies the software items. It defines key responsibilities, external properties, relationships among components, and architectures for internal and external interfaces.
14. How does IEC 62304 address the use and documentation of Software of Unknown Provenance (SOUP)?
SOUP (Software of Unknown Provenance) must be uniquely identified by title, manufacturer, and unique designator (e.g., version or patch number). Manufacturers must specify its required functional/performance characteristics and supporting hardware/software. Risks related to SOUP failure must be analyzed, specifically evaluating published anomaly lists.
15. Is the Detailed Design required for all the Software Items?
No, the full detailed design documentation (including documenting design for each software unit and interfaces, and verifying the design) is mandatory only for Software Items classified as Class C. Software is only subdivided into software units for Classes B and C.
Software Unit Implementation and Verification [Clause 5.5]
16. What is a Software Item?
A software item is any identifiable part of a computer program (source code, object code, control data, or a collection of these). It is a general term encompassing components at all levels of decomposition, from the top-level software system down to the lowest-level software unit.
17. Can I use SOUP in a Medical Device?
Yes, SOUP can be incorporated into a medical device. However, the manufacturer takes responsibility for the acquired component (SOUP) and must include it in the overall risk management of the medical device. Its integration and risks must be addressed in the software development plan.
18. What is the difference among SOUP, OTS and COTS?
SOUP (Software of Unknown Provenance) is a software item already developed and generally available but not developed for incorporation into the medical device; they are also known as OTS. SOUP is also defined as an item previously developed for which adequate records of the development processes are not available. The latest part of the definition applies also to items previously developed by the same manufacturer. The COTS (Commercial Off-The-Shelf) differ from OTS when they are made available by commercial suppliers.
Software Testing [Clause 5.6, 5.7]
19. What is the difference between Software System Testing and Validation?
Software System Testing verifies the software’s functionality, confirming all specified software requirements have been implemented. Validation involves demonstrating that the overall medical device meets its intended use requirements. Validation and final release of the medical device are outside the scope of IEC 62304.
20. How should test results and overall verification status be summarized in the Software Test Report?
The report must document the test result (pass/fail and a list of anomalies) found, along with a reference to the test case procedures showing expected results. It must also record the version of software tested, relevant hardware/software test configurations, test tools used, the date tested, and the identity of the tester.
Software Release [Clause 5.8]
21. Is the Software Release different from the Product release?
Yes. Software release (Clause 5.8) is the process of making the software available for utilization at a system level. However, this standard does not cover validation and final release of the medical device itself.
22. Does the IEC 62304 indicate the essential information that must be documented in the Software Release Note to support device submission?
Yes, the IEC 62304 defines the essential information to be included in the Software Release Note for Medical Device submission, even though it does not mandate a specific document format. The manufacturer must document the released version, all known residual anomalies, and the procedure/environment used to create the released software (for Classes B/C). Documentation archives must be retained for the longer of the device life or the time specified by relevant regulatory requirements.
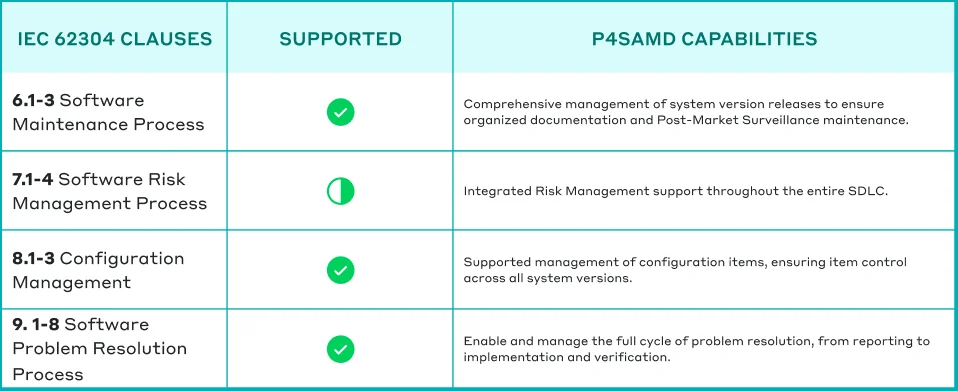
Software Maintenance and Problem Resolution Process [Clause 6, 9]
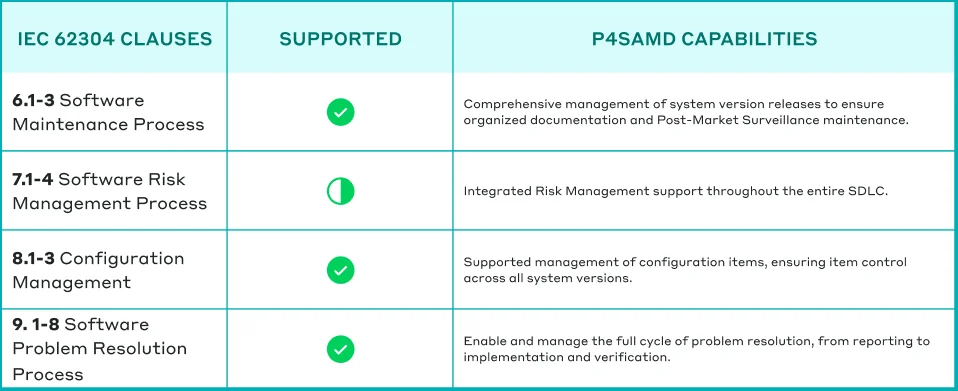
23. What is the key difference between the Software Maintenance Process (Clause 6) and the Software Problem Resolution Process (Clause 9)?
Maintenance (Clause 6) focuses on the overall adequate response to feedback arising after release, managing high-level decisions regarding safety impacts and ensuring compliance with local regulations. Problem Resolution (Clause 9) is the internal control system (defect tracking) used by the maintenance process to analyze specific problem reports and generate necessary change requests.
24. How does the standard govern the process for capturing, assessing, and resolving post-market Software Problem Reports?
The standard defines the Problem Resolution process to address the possible software defects. Feedback must be monitored and documented as a problem report. The report must be evaluated for its relevance to safety using the software risk management process. Resolution uses the change control process (8.2) to approve/implement change requests, and the manufacturer must verify resolutions and advise relevant parties.
Configuration Management [Clause 8]
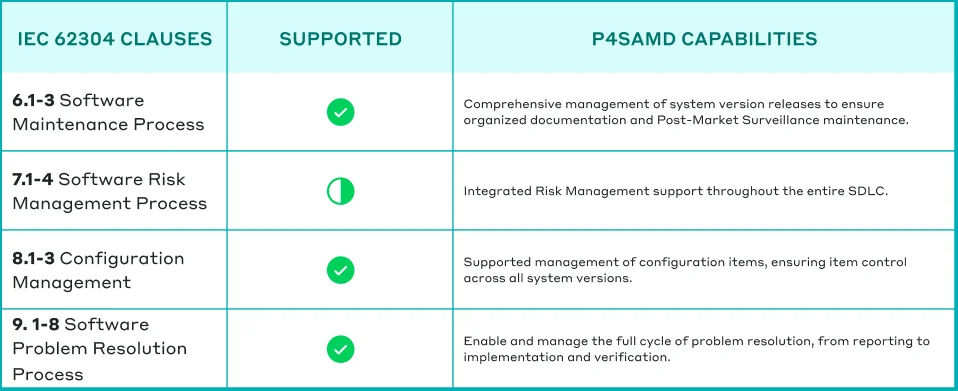
25. What is a “Configuration Item”?
A configuration item is an entity that can be clearly identified and tracked at a specific point in time. It can represent all elements needed to build your software, such as build files, source code, compiler settings and documentation. Essentially, any key item for creating the software counts as a configuration item.
26. Why are identifying and managing the Configuration Items important?
Identifying configuration items and their versions is necessary to identify which parts comprise the software system configuration. Configuration management (Clause 8) is vital to recreate a software item, identify its constituent parts, and provide a history of the changes made to it throughout the life cycle.
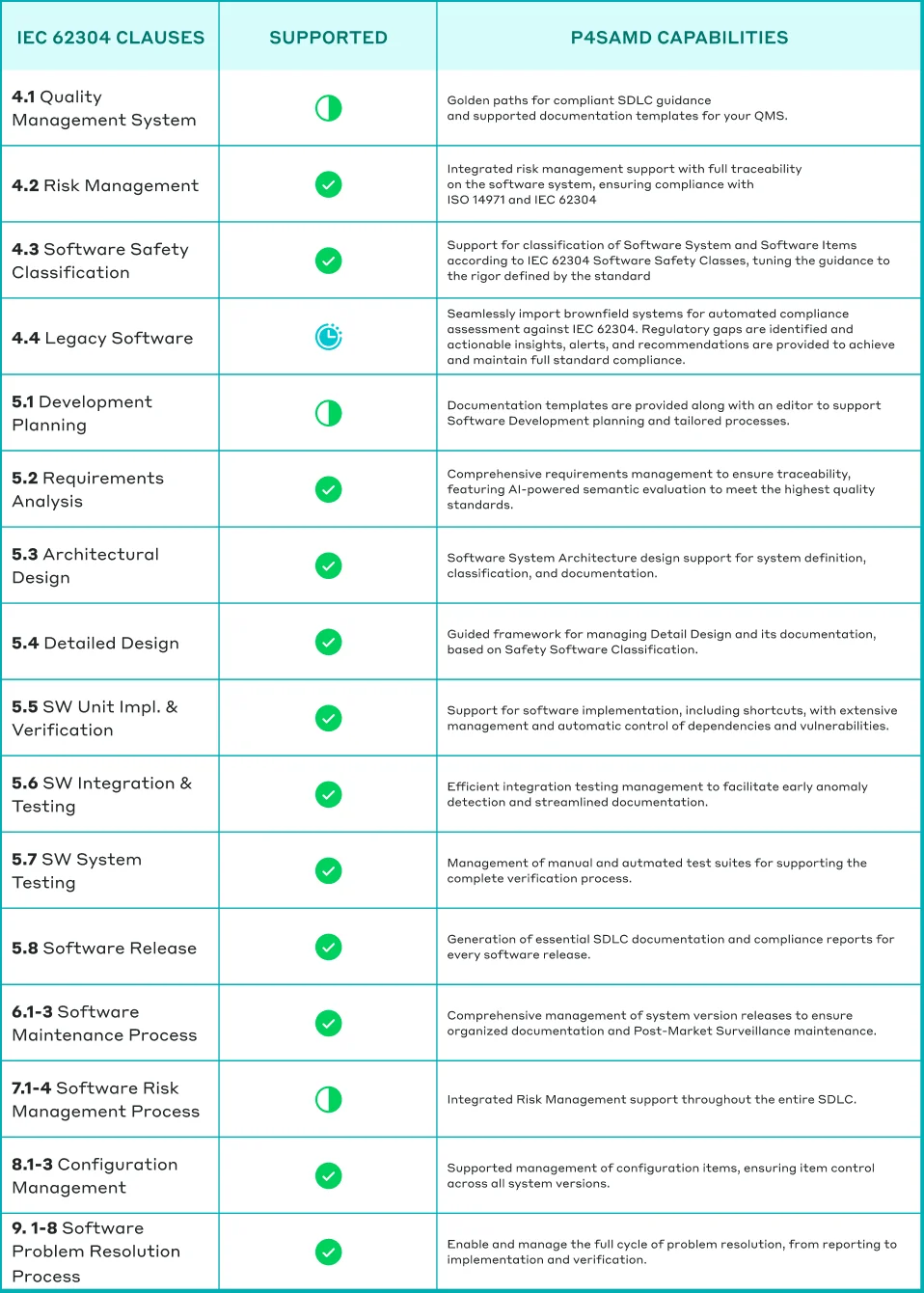
Table: How P4SaMD supports comprehensive adherence to IEC 62304.